

BioByte 018: a biotech love poem, errors in high-impact genetics studies, human-level AI timelines, intratumor microbiota
BioByte 018: a biotech love poem, errors in high-impact genetics studies, human-level AI timelines, intratumor microbiota




Welcome to Decoding Bio, a writing collective focused on the latest scientific advancements, news, and people building at the intersection of tech x bio. If you’d like to connect or collaborate, please shoot us a note here. Happy decoding!

Happy Valentine’s Day bio fam! In honor of the holiday, here’s a biotech love poem courtesy of ChatGPT:
Biotech, oh Biotech, my heart's delight,
In you, I find a love that's just right,
Your science and innovation, so divine,
Makes my heart beat faster, oh how you shine.From DNA sequencing to gene editing,
You are a true marvel, ever so intriguing,
Your research and development, a sight to see,
Like a beautiful symphony, so sweet and lovely.Your advancements in medicine, oh so grand,
You cure diseases and save lives, just as planned,
You are the epitome of innovation and growth,
A love that blossoms, like a beautiful rose.Oh, Biotech, my love for you will never fade,
With you by my side, I'm never afraid,
You are the future, the hope, the dream,
My heart beats for you, oh my Biotech queen.
Big week in bio. Let’s get after it:

What we read
Blogs
Highly cited genetics studies found to contain sequence errors [Kwon, Nature News 2023]
This article explores the prevalence of mistakes in published gene research based on a comprehensive literature review of high impact journals: Molecular Cancer and Oncogene.
These efforts build upon past work of Byrne and team after reviewing over 12,000 papers using Seek & Blastn, software that extracts short nucleotide sequences mentioned in papers, spots potential errors, and cross-checks those against a public database of nucleotides known as BLASTn. Ultimately, the researchers found >700 papers citing experimental reagents that had errors in their DNA or RNA sequences.
After screening reagents purported to target unmodified human genes or genomic sequences in >330 papers, the team found errors in 253 (~4%) of papers analyzed. In the discussion, the authors acknowledge that some of the errors identified could have been unintentional, and some errors were unlikely to have a significant effect on the conclusions of the papers.
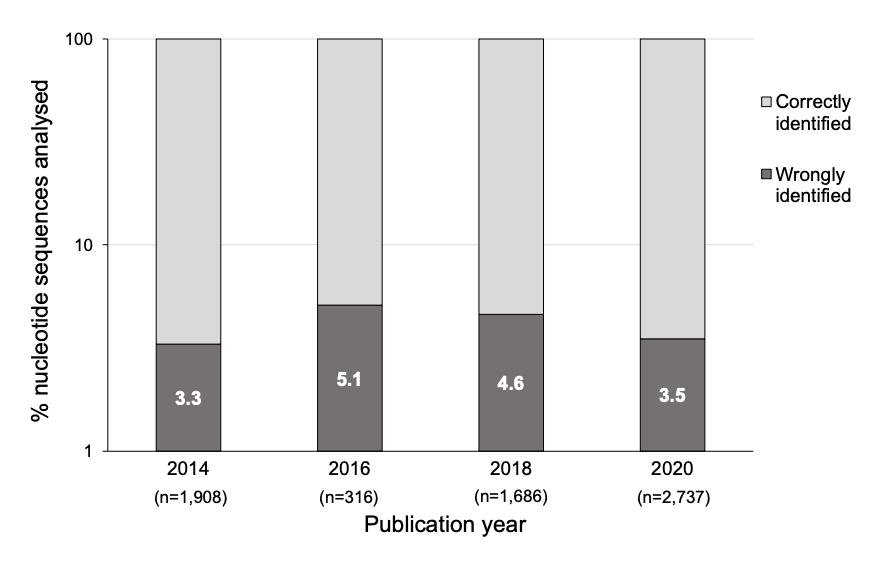
Interestingly, about 25% of the Oncogene papers with errors have also been flagged on the post-publication peer-review platform PubPeer, but mostly for separate, image-integrity issues. Here is a link to the pre-print.
AI timelines: What do experts in artificial intelligence expect for the future? [Roser, Our World in Data, February 2023]
In this brilliant post on Our World in Data, Roser grapples with key questions such as: “Does Human-level Artificial Intelligence Exist?” using online forecasting platforms such as Metaculus. The article defines human-level AI as “a machine, or a network of machines, capable of carrying out the same range of tasks that we humans are capable of.”

The two big takeaways are:
There is no consensus, and uncertainty is high, even among experts.
There is general agreement in the overall picture, and that the timelines are shorter than a century.
“The majority of those who study this question believe that there is a 50% chance that transformative AI systems will be developed within the next 50 years. In this case it would plausibly be the biggest transformation in the lifetime of our children, or even in our own lifetime.”
Cancer debugged [Monique Brouillette, Nature Biotech, 2023]
Why it matters: Tumors often possess characteristic microbiota, which may serve as novel diagnostic or therapeutic biomarkers. The presence and role of intratumoral bacteria is a promising emerging area of precision oncology. Since a landmark Science paper in 2017 which discovered that bacteria within pancreatic adenocarcinoma produce an enzyme that can inactivate a chemotherapeutic agent contributing to treatment resistance, there’s been a flurry of papers demonstrating associations between microbes and various cancers. Some of the findings have been controversial, and skeptics have raised concerns about the possibility of contamination (no surface is safe from bacteria in the laboratory), although a well-controlled follow-up study in 2020 discovered bacteria inside tumor cells.
Despite the potential for intratumor bacteria in cancer, links between the gut microbiome and cancer are better understood and closer to clinical translation. There are a number of diagnostic companies commercializing microbiome-based tests, such as BiomeDx and Metabiomics
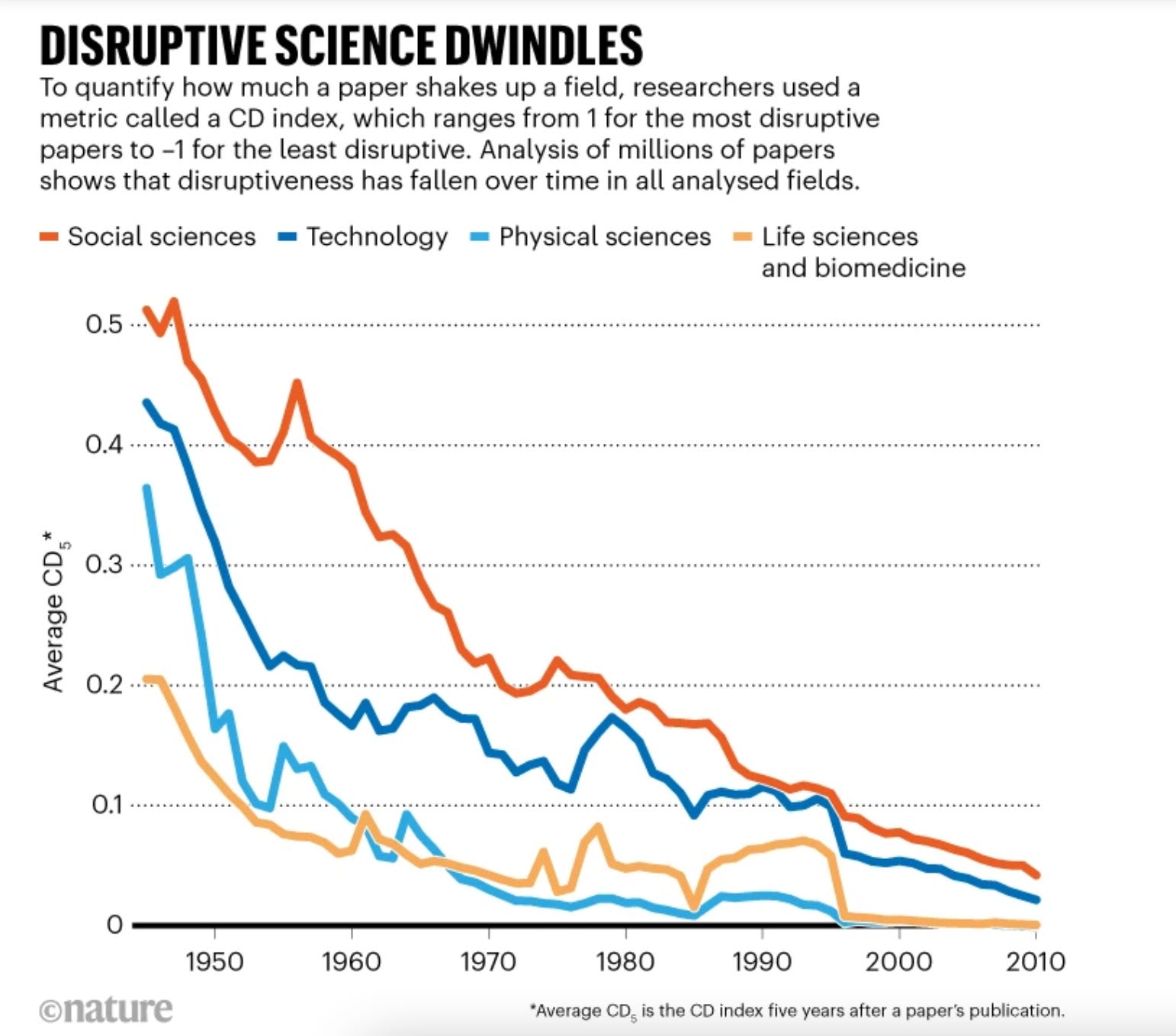
Disruptive Science has Declined and No One Knows Why [Nature News, 2023]
Despite the number of published scientific research increasing in the last few decades, shockingly few of them have been “disruptive” or otherwise creating a step-change from previous literature.
“You don’t have quite the same intensity of breakthrough discoveries you once had”
This study dove into citations as a metric for disruptiveness—how often the study itself was cited as opposed to the references in that study. While there are several confounding variables at play such as team size, incentives, and language around communicating scientific discoveries, the trends suggest some caliber of reform may help bolster scientific rigor.
Academic papers
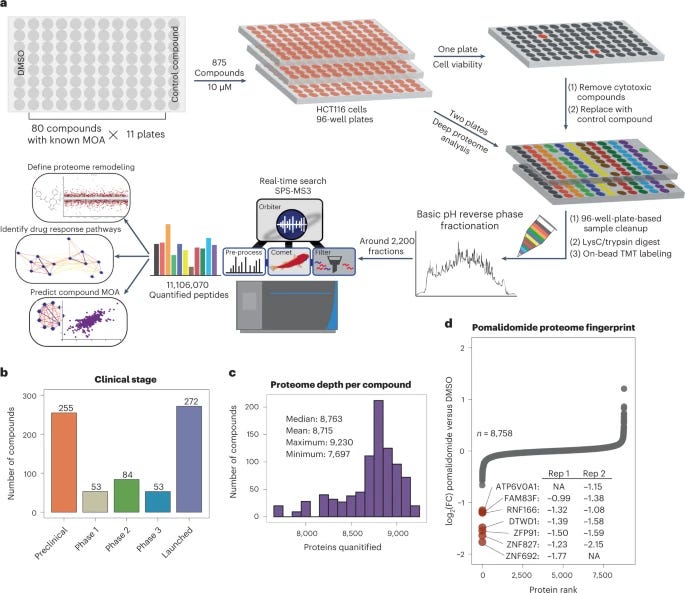
A proteome-wide atlas of drug mechanism of action [Mitchell et al., Nature Biotechnology, 2023]
Why it matters: systems-level profiling is required to elucidate the mechanism of action (MOA) of a drug. Most approved drugs act via interaction with protein targets, yet high-throughput methods to identify protein-compound interactions have not achieved comprehensive proteome depth. In fact, no resource of small-molecule induced changes in protein expression have screened more than 80 compounds. The authors of this study quantified proteome fingerprints for 875 small molecules, 10x the previous largest screen. This high-throughput, deep proteome profiling method can be used for better MOA deconvolution and compound repurposing.In this elegant study, Mitchell et al. showcase the largest isobaric labeling study to date, and the first deployment of isobaric-labeling-enabled proteome profiling in 96-well-plate-based cellular screening.
The authors of the paper performed a high-throughput proteomic screen using 875 small molecule compounds and a human cancer cell line (HCT116). To quantify the amount of protein after the cell was screened with one of the compounds, the researchers used tandem mass tags (TMTs) which are a form of ‘isobaric labeling’ tags. These TMTs bind to peptides present in the wells. TMTs belong to a family of chemicals that have the same mass “but yield reporter ions of differing mass after fragmentation”, and have historically allowed increased throughout in proteomics. This type of labeling allowed deep proteome profiling and the inference of new polypharmacology and MOAs for small molecules, some of which are currently in the clinic.
The paper has many take aways and is worth reading in detail, but here are some excerpts on the findings:
“We provide the largest resource for evaluating the frequency and specificity of ligand-induced changes in target protein regulation, demonstrating that 15% of compounds modulate their target protein’s abundance.
We used the depth of our screen to appreciate polypharmacology of the FDA-approved poly (ADP-ribose) polymerase (PARP) inhibitor talazoparib, revealing off-target proteins with high confidence.
We show that many widely used tool compounds and clinically relevant small molecule inhibitors have myriad unanticipated proteome-level effects. Analysis of these unexpectedly active compounds uncovered a family of structurally related small molecules, including PAC-1, currently in clinical trials, whose activity is driven by iron chelation.
Finally, we highlight the activity of JP1302—an adrenergic receptor antagonist that inhibits transcription through several mechanisms including degradation of linker histone H1.”
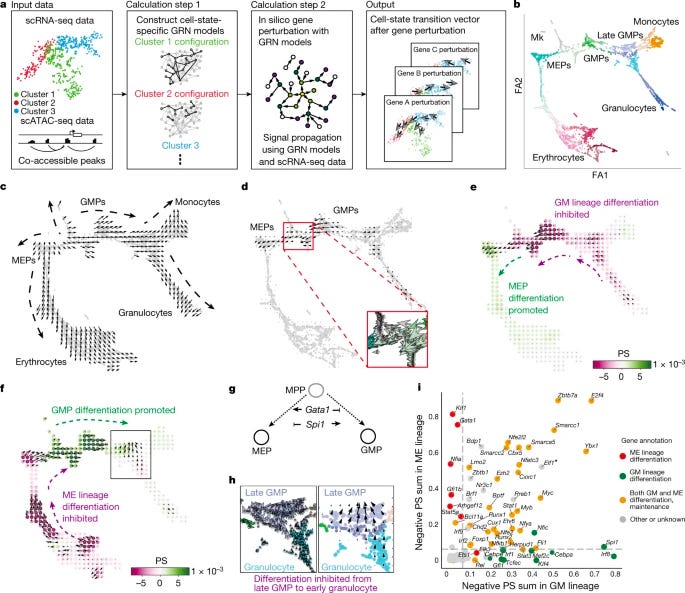
Dissecting cell identity via network inference and in silico gene perturbation [Kamimoto et al., Nature, February 2023]
Why it matters: Single-cell methods have shown immense promise in characterizing cell identity. Computational approaches for simulating single cell phenotypes after perturbation are emerging, though have historically required experimental perturbation data for model training, which presents limitations for scalability. This method overcomes these limitations by integrating mulutimodal data to build custom gene-regulatory network models that simulate shifts in cell identity after transcription factor perturbation without requiring experiemnetal perturbation data. Applications for this technology are vast, and show promise for understanding cell state transformation for mapping the etiology and progression of disease.Cell identity is governed by complex systems of gene expression, which are known as gene-regulatory networks. This paper by Kamimoto et al. inferred gene-regulatory networks from single-cell multi-omics data to perform in silico transcription factor perturbations in order to understand changes in cell identity in mouse and human hematopoeisis and zebrafish embryogenesis using a machine-learning based approach called CellOracle (Github). CellOracle was able to simulate shifts in cell identity via interpretation of systematic gene-to-gene relationships for various cell states, and demonstrataed an ability to identify previously unreported phenotypes including early-stage-specific cell-fate regulatory genes such as Tal1 in erythropoeisis.
Advancing CAR T cell therapy through the use of multidimensional omics data [Yang et al., Nature Reviews Clinical Oncology, 2023]
Why it matters: CAR-T cell therapy has revolutionized the treatment of B cell malignancies, yet important challenges remain. Multi-omic analyses will be beneficial in addressing important bottlenecks in cell therapy including extending durability of therapeutic response, improving response rates, reducing toxicity, and targeting solid tumors. A comprehensive review was recently published in Nature Reviews Clinical Oncology on how multi-omics data such as genomics, epigenomics, transcriptomics, TCR-repertoire profiling, and other modalities can aid in the engineering of CAR-T therapies. The review is worth a full read, but some highlights:
Multi-omic data has been valuable in the identification of optimal targets for highly specific CAR-T therapy. For example, comparative analyses of genomics, transcriptomics, and proteomics from tumor and non-tumor tissue has facilitated the discovery of novel targets, as well as engineering combinatorial CAR targets (figure below).
An important challenge in CAR-T therapy is T cell exhaustion - a state of dysfunction that can occur in T cells due to persistent activation, resulting in decreased tumor killing ability. Multi-omic analyses are helping identify molecular markers and predictors of T cell exhaustion, such as the identification of the transcription factor BATF as a key mediator in countering T cell exhaustion and pushing T cells towards an effector phenotype.
Cytokine release storm (CRS) results from cytokine production by CAR-T cells following engagement with target cells as well as from endogenous immune cells, and is the most serious side effect of CAR-T therapy. Bulk and single-cell RNA-seq as well proteomic studies have converged on a set of cytokines and receptors (namely IL-6 and IL-1) that most likely mediate CRS, and may serve as potential therapeutic targets.

What we listened to
The power of Bayesian Statistics [BBC, The Life Scientific]
Imogen Pryce, COO of R&D at Relay Therapeutics [LifeSci Beat]
Notable Deals
With $520 million in hand, two entrepreneurs try to upend the biotech funding model [STAT]
Cerevance Expands Series B Financing with Additional $51 Million
As clinical trials get more complex, software startup Faro bags $20M to help visualize them [Endpoints]
Garuda Therapeutics Raises $62M in Series B Financing
In case you missed it

What we liked on Twitter








Events




Field Trip


Did we miss anything? Would you like to contribute to Decoding Bio by writing a guest post? Drop us a note here or chat with us on Twitter: @ameekapadia @ketanyerneni @morgancheatham @pablolubroth @patricksmalone