

BioByte 052: supercomputers and comp bio, foundation models for genes and cells, neural scaling of deep chemical models, ML-driven T cell infiltration prediction for solid tumors
BioByte 052: supercomputers and comp bio, foundation models for genes and cells, neural scaling of deep chemical models, ML-driven T cell infiltration prediction for solid tumors




Welcome to Decoding Bio, a writing collective focused on the latest scientific advancements, news, and people building at the intersection of tech x bio. If you’d like to connect or collaborate, please shoot us a note here or chat with us on Twitter: @ameekapadia @ketanyerneni @morgancheatham @pablolubroth @patricksmalone @zahrakhwaja. Happy decoding!

Your weekly dose of AI x bio, delivered to your inbox! Have you checked out our newly revamped “Notable Deals” section? 🔥🔥🔥
The Decoding Bio team is working on a feature project highlighting new lab launches by up-and-coming PIs. To nominate a lab or PI, please contact us via email (decodingbio@gmail.com), Twitter DMs, or smoke signal.
Happy Decoding!

What we read
Blogs
AI in DD — where are the success stories? [Leo Wossnig, October 2023]
Leo Wossnig, CTO of LabGenius, published a piece this week responding to the flurry of recent commentaries on the status of AI-driven drug discovery. The essay is packed with insight, but one point is worth expanding on:
“Knowing what to optimise for is most often the biggest question. Sometimes we know what we’d like but don’t know how to optimise for it, or (more often) we simply do not yet know what to optimise for since we don’t have a good understanding of disease biology!”
In machine learning, one of the most important methodological choices is selecting an objective function (aka cost or loss function). The objective function quantifies how far off a prediction is from the actual result, and is optimized through model training. With a clear and task-relevant objective function, and sufficient high-quality training data, ML is extremely effective.
The challenge in computational biology and drug discovery, however, is that the right objective function is often not clear. Biological systems are complex, and we have incomplete information. As drug hunters, the overarching objective is known ("develop safe and effective drug-like molecules"), but translating this goal into granular, quantifiable objective functions for ML is super challenging.
Let's say the goal is to develop an antibody therapeutic. One reasonable objective function is increasing binding affinity of the antibody to the therapeutic target. We've developed really effective ML models (protein language models, diffusion models, etc) for designing strong antibody binders. But biology is more complicated, and turns out that for many therapeutic applications, binding isn't the most important variable for a safe and effective drug. Earlier this year, a paper was published in Nature (Yu et al., Feb 2023) that showed that when designing antibodies antagonists to PD-1 (the most important receptor in immuno-oncology), *low* affinity binders, not strong binders, resulted in more potent signaling and effects on T cells. ML would have successfully optimized the specified objective function, but given the complexity and unknown unknowns of biology, still wouldn't have developed a successful drug.
An additional issue is that in drug discovery, multiple objectives require co-optimization. The example of antibody drugs is helpful here too. In 2017, a landmark paper was published in PNAS that showed that the most important factors that predict the success of antibody candidates in clinical trials isn't only how well the antibody binds to the target, but a bunch of biophysical properties of the antibody (collectively termed developability parameters) such as stability and immunogenicity. The problem for computational biology is that all of these parameters need to be co-optimized, and often inversely correlate. For example, a stronger binder often leads to increased risk of immunogenicity. ML excels at multi-parameter optimization, so is well-suited for this problem. But the issue of thoroughly specifying all relevant objective functions remains.
To summarize, one of the most important bottlenecks in AI-driven drug discovery is the thoughtful and systematic selection of multiple objective functions for the ML model, balancing trade-offs between each of these variables, and aligning the objectives with the desired therapeutic result.
Analysis of pharma R&D productivity a new perspective needed [Schuhmacher et al., Drug Discovery Today, October 2023]
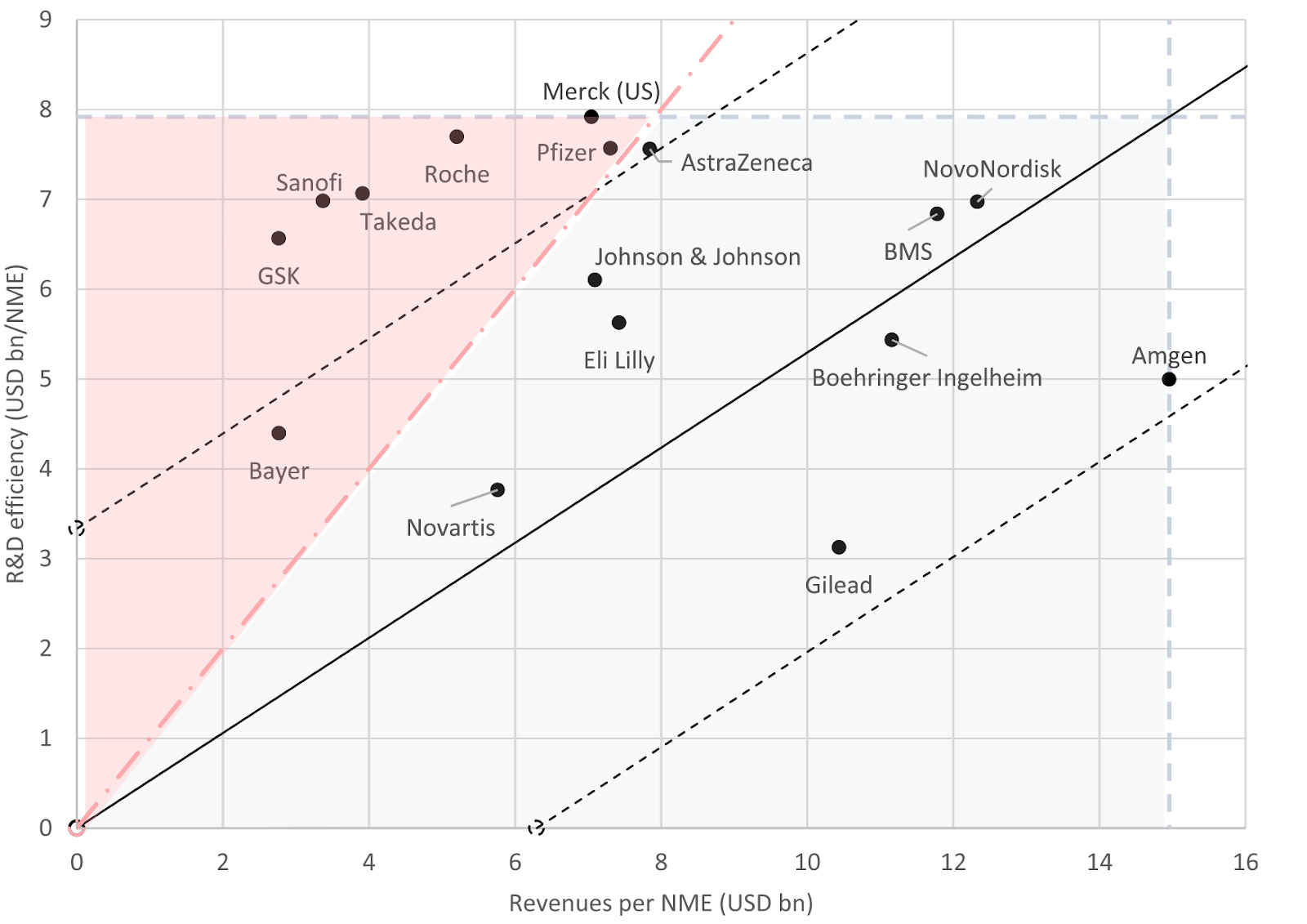
This feature explores R&D productivity and efficiency of 16 big pharmaceutical companies over a 20 year period (2001-2020).
Setting the scene:
Industry’s R&D efficiency is recovering due better understanding of disease biology, improved target selection and validation, PK/PD modeling and patient stratification.
The high number of approvals by the CDER of the FDA in 2020-2021 suggest that R&D efficiency is not declining further
R&D input of big pharma: in 2020, their cumulative sales were $511B (62% of the total 2020 prescription market). In those 20 years, they invested $1.54T in R&D.
R&D output of big pharma: 251 new therapies were approved in the 20 year period (46% of all therapies). There was an increase in the average annual output of 9 new drugs per year in the first decade to 15.7 new drugs per year in the second decade; yet a declining relative proportion of FDA-approved drugs by big pharma from 76% (2001) to 25% (2020)
R&D efficiency and productivity of big pharma:
The authors used the formula: R&D efficiency of company = annual R&D spend / number of new drug launches
The group’s R&D efficiency in the 20 year period is $6.16B; double the estimated pre-approval R&D costs per NME ($2.6B).
The R&D effectiveness (commercial outcome per new drug) of big pharma was on average $7.57B per new drug.
R&D productivity was classified by plotting R&D efficiency vs new drugs launched in the 20 year period (image above). The red area indicates the companies that have a negative R&D productivity. Most of which compensated for this via M&A of previously approved drugs except Takeda.
Will Supercomputers and AI Drive Biotech Breakthroughs? [Chatsko, Living Tech, October 2023]
The symbiotic relationship between supercomputers and AI in propelling biotechnology forward is under the spotlight. The biotech sector is inundated with vast amounts of intricate data that demands unprecedented computing prowess. Supercomputers, such as Anton 2, have emerged as instrumental in simulating prolonged protein motions, thereby aiding in drug design. Concurrently, the prowess of AI algorithms, notably AlphaFold 2, in predicting protein structures has taken a leap. Yet, the intricate tapestry of biology implies that AI and machine learning models are in dire need of expansive and meticulously annotated datasets, which are currently in short supply.
While neuroscience is poised to leverage the strengths of these advanced computational tools, the biomanufacturing realm is still grappling with data acquisition and inter-sector collaboration. The bridging of biotech trailblazers like Recursion and Ginkgo with tech behemoths such as Nvidia and Google Cloud heralds the dawn of "foundation models" in biology, potentially akin to ChatGPT's proficiency in protein structures. Both specialized supercomputers, like Anton 2, and versatile systems, epitomized by BioHive-1, are catalyzing simulations in biology to hasten research trajectories.
The consensus among experts is unambiguous: the harmonious fusion of supercomputers and AI can be the linchpin for monumental strides in biotech, provided the impediments of data access and utilization are navigated deftly, potentially through innovative partnerships and a recalibrated mindset.
The Isoform Frontier [Simon Barnett, October 2023]
Gene expression profiling is undergoing a renaissance with the ability to sequence full-length mRNA isoforms cost-effectively at scale. Isoforms result from mechanisms like alternative splicing and contain different combinations of exons that translate into distinct proteins. Analyzing isoforms will provide new insights into functional genomics, biomarkers, and disease.

RNA-seq is limited in studying isoforms since its short reads cannot span long isoform transcripts. New long-read sequencing from PacBio and Nanopore with high accuracy, throughput, long reads, and low costs now enable precise isoform sequencing. Adoption of these platforms is growing rapidly.
PacBio’s new Revio platform and MAS-Seq chemistry boost throughput and lower costs. Nanopore’s new pore designs and basecalling software increase raw read accuracy above 99%. Together these breakthroughs satisfy the criteria needed for ubiquitous isoform sequencing.
An exciting application is combining isoform sequencing with mass spectrometry proteomics. Isoforms improve protein identification by providing sample-specific databases. A recent study found that isoform sequencing identified many novel protein variants and changed proteoform classifications. Though limitations exist, this workflow is poised to impact proteomics given existing adoption of both techniques.
Simon predicts long-read sequencing will continue rapid adoption, catalyzing genome annotation, biomarker discovery, and research across many disease areas as isoform analyses become standard practice.
Academic papers
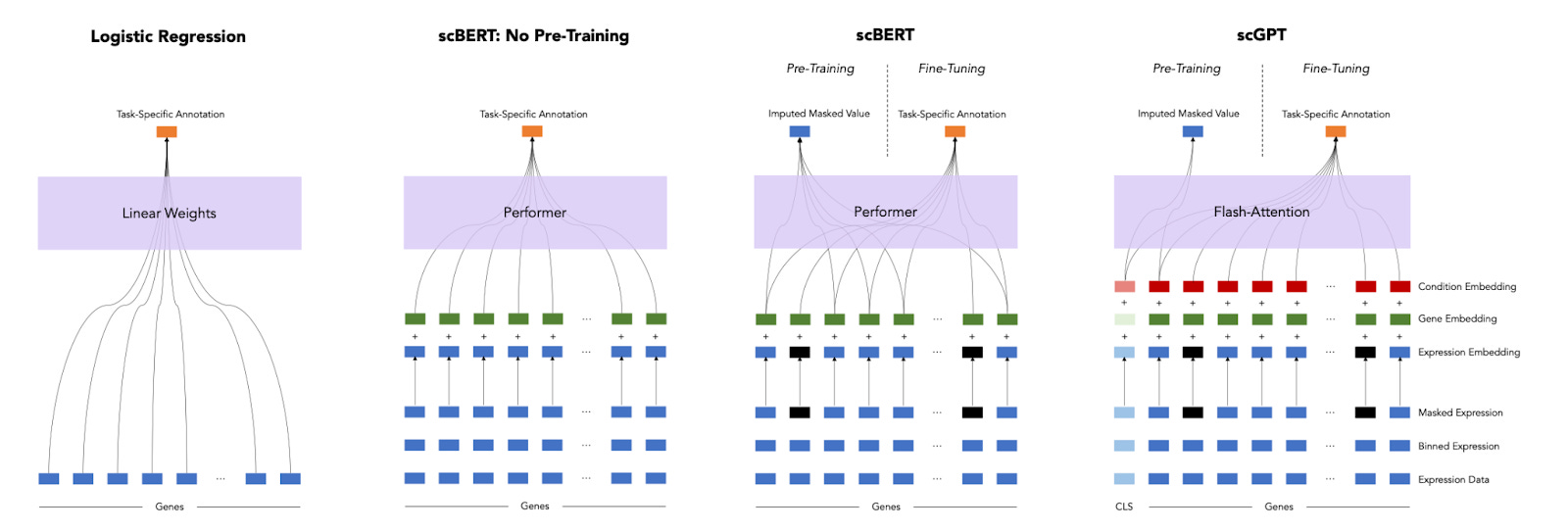
A Deep Dive into Single-Cell RNA Sequencing Foundation Model [Boiarsky et al., BioRxiv, Oct 2023]
Why it matters: In a new preprint, the performance of foundation models for single cell RNA-seq applications was found to be similar to simpler linear models, highlighting the importance of baselines and benchmarks when evaluating advanced, non-linear models for biological applications.Given all of the recent developments in foundation models for single cell RNA-seq applications, it is important to ask: how much better are these models than simpler, linear models? The answer according to a new preprint is: not much, at least for the tasks tested. A simple logistic regression model performed as well as foundation models like scGPT and scBERT on a cell annotation task for most cell types. Cell annotation is likely not a challenging enough task to demonstrate the value of pre-trained models, but a more thorough and systematic evaluation of what type of tasks are best suited to foundation models is needed.
Neural scaling of deep chemical models [Frey et al, Nature Machine Intelligence, October 2023]
Why It Matters: The neural scaling laws provide practical guidance for efficient model development in chemistry. The results show that unlike other domains, extremely large models and datasets are needed to minimize loss and reach potential performance limits. Methods like TPE make large experiments more feasible. Overall, the work demonstrates the promise of scale in chemical deep learning and provides a framework for further research.This research investigates how increasing model size and data size impacts performance for deep learning models in chemistry. Two types of models are studied - large language models for molecules and graph neural networks for molecular dynamics. The goal is to discover neural scaling laws that relate resources like model size and data to model loss.
Large generative language models called ChemGPT with up to 1 billion parameters are trained on up to 10 million molecules from PubChem. Surprisingly, model loss keeps decreasing with larger models and data sizes. Power-law scaling relations are identified, with exponents showing the efficiency of scaling.

Graph neural networks are trained to predict molecular properties using up to 100,000 quantum chemistry calculations. Increasing model capacity and data size improves results, especially for models with physics-based inductive biases. Equivariant models show greater efficiency in the scaling laws.
To enable large experiments, a method called training performance estimation (TPE) is introduced to accelerate hyperparameter tuning. TPE identifies optimal settings using only a fraction of the compute budget.
Generating counterfactual explanations of tumor spatial proteomes to discover effective strategies for enhancing immune infiltration [Wang et al., aRxiv, 2023]
Why it matters: Targeting solid tumors through immunotherapies has been challenging due to the tumor microenvironment (TME) as it alters the immune composition around the tumor. The ability to predict which perturbations could increase T-cell infiltration could enable companies to design better cancer therapeutics.T-cell infiltration is required for immunotherapies, such as CAR-Ts and checkpoint inhibitors, to be effective. This has been a challenge in solid tumors due to the TME. The interactions between tumor cells and immune cells can alter the composition of which types of immune cells are available. For example, high levels of TILs in the TME are associated with improved prognosis, whereas low levels of TILs are associated with poor prognosis and reduced response to immunotherapy.
Spatial omics techniques capture cellular organization and activity in human tumors with unprecedented molecular detail; revealing relationships between signals, TME composition and potentially drug response.
The authors formulated the T-cell infiltration prediction as a ML problem. They trained a convolutional neural network to predict T-cell level based on spatial images of the signaling molecules in the TME. They then computed perturbations to the signaling molecules that the network predicted could increase T-cell abundance.
When applied to melanoma, their approach predicted 5 chemo- and cytokine perturbations that can convert immune-excluded tumors into immune-inflamed. The networks were also applied to CRC liver metastases and breast cancer.
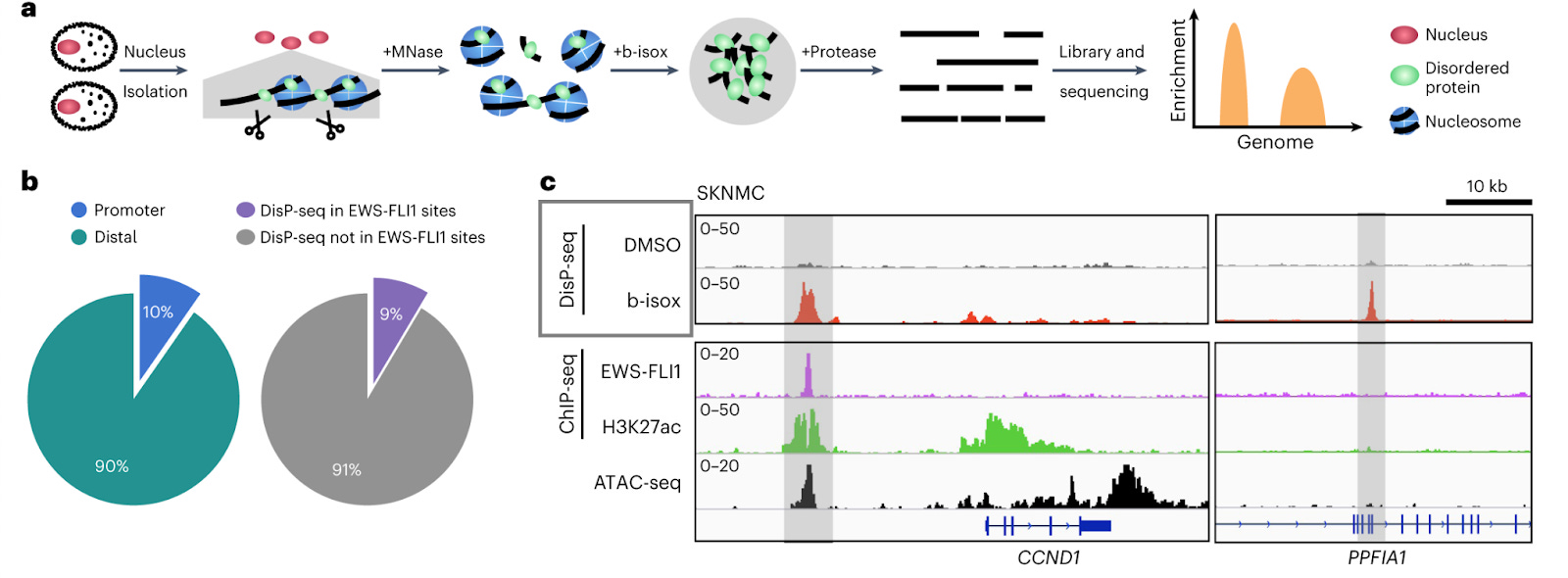
DisP-seq reveals the genome-wide functional organization of DNA-associated disordered proteins [Xing et al., Nature Biotechnology, 2023]
Why it matters: Many proteins (such as transcription factors) contain Intrinsically disordered regions (IDRs) – areas which lack a fixed three-dimensional structure – are known to play an important role in gene regulation. However, we still lack a sophisticated understanding of their distribution and impact across the genome. In this paper, the authors develop DIsP-seq (disordered protein precipitation followed by DNA sequencing), allowing them to decipher the distribution and function of IDRs.To date, technologies for probing DNA-binding IDRs use antibodies to precipitate proteins; this generally requires the addition of an engineered tag, or working with those that are well studied and have known binders. IDRs are fluid and transition across several conformations - making their functional characterization challenging. Here, the authors use biotinylated isoxazole (binds to proteins with low-complexity regions - regions of biased composition, containing simple sequence repeats) to selectively precipitate IDR-containing proteins bound to DNA. They then treat the samples with nuclease to fragment unprotected DNA, and also use proteinase K, and RNase before massively parallel sequencing and mapping the reads to the genome. This readily uncovers the distribution of IDRs across the genome, which can then help identify binding motifs and enriched regions. The authors used DisP-Seq to probe the oncogenic fusion protein EWS-FLI1 (in which the IDR from ESWR1 is fused to the DNA-binding domain of the transcription factor FLI1) in Ewing Sarcoma, and demonstrate that cell-type specific spatial organization of DNA-binding IDR-containing proteins can significantly impact pathologic gene regulatory dynamics.
Although DisP-seq cannot link detection of DNA regions with discovery of novel IDR-containing DNA-associated proteins (necessitates downstream techniques, such as mass spec to identify precipitated proteins), it is a quick and cost-effective means to study how DNA-associated disordered proteins are distributed across the genome.
GenePT: A simple but Hard-to-Beat Foundation Model for Genes and Cells Built from ChatGPT [Chen and Zou, BioRxiv, October 2023]
Why it matters: GenePT shows that language model embeddings of text summaries can effectively capture the underlying biology of genes and cells, representing a simple and efficient alternative to existing methods requiring extensive data curation and pre-training. This work opens up new avenues for transfer learning in biology using common sense and literature within language models like ChatGPT. Combining these embeddings with specialized learned representations may provide additional gains. The accessibility of GenePT could help democratize computational biology and lower the barriers to utilizing modern NLP techniques.GenePT offers a novel approach to gene representation by harnessing summary texts from renowned databases such as NCBI, integrated seamlessly with ChatGPT embeddings. Delving deeper into the methodology, genes are depicted through two distinct processes: by averaging the gene vectors grounded on expression, known as GenePT-w, or by embedding the hierarchically arranged gene names in a structured sentence form, termed GenePT-s. Impressively, even without any preliminary dataset training, GenePT has showcased its prowess by either equating or outperforming recent niche models, including Geneformer and scGPT. This superiority is evident in a myriad of tasks, ranging from predicting gene attributes to discerning gene-gene interactions and annotating cell types.
Specifically focusing on gene-centric tasks, GenePT, when coupled with a rudimentary classifier, boasts a staggering 95% accuracy in forecasting functionality classes, spanning across a spectrum of 15 diverse categories. It further clinches an AUC of 0.84 in anticipating gene-gene interactions, a feat that transcends previous benchmarks. Transitioning to the realm of cell annotation, GenePT-s mirrors the efficiency of scGPT. Interestingly, amalgamating the strengths of both models amplifies accuracy, hinting at the potential symbiotic nature of the information they each possess. A salient feature of GenePT is its resilience against batch discrepancies while steadfastly preserving crucial biological indicators associated with disease manifestations and cell types.
Notable Deals
Roche / Roivant: A $7.1B buyout causes a stock tumble - why?
Roivant: Holding company for a number of specialised biotech entities (“vants”). Each focuses on the development of a particular tech or asset.
Telavant: A Roivant subsidiary set up to house RVT-3101 - a TL1A antibody acquired in Dec 2022 from Pfizer for $45M upfront (Japan and US rights). Telavant positioned the asset for phase III with $50M in expenses.
Deal Overview:
Roche will acquire the rights to develop and manufacture RVT-3103 in the US and Japan (Pfizer keeps ROW) for $7.1B (+ $150M milestones on Phase III initiation) one year later. This creates approx $5.2B in value for Roivant (Roivant owns 75% of Telavant and Pfizer retains 25% stake).
Rationale for Roche: TL1A fever: Large market (estimated $15B for IBD alone in the US), potential application in several other immune conditions. Analyst estimated peak sales for RVT-3103 is $4.5B by 2029. Sanofi and Merck caught the fever too.
Why did the stock drop?
At face value this looks like rapid value creation and validation of a risky biotech business model, in which the rich proceeds combined with ripe BD market could lead to the build up of a valuable pipeline. However, this transaction gives away a Phase-III asset with strong data in ulcerative colitis (subset of IBD) which was the main attraction to the stock for many investors.
Interesting business model:
Roivant’s business model works by sifting through thousands of shelved / developing assets at pharma and identifying those that, with additional development, have blockbuster potential. A core requirement is that these assets have evangelists within the pharma. The model is essentially a risk-sharing arrangement with pharma for assets they may not have the bandwidth to pursue.
Antibody-drug-conjugates (ADC): An exploding modality
ADCs are essentially a form of targeted chemotherapy - the chemotherapy is attached to an antibody to provide specificity. Once a dismissed modality, the field has recently exploded due to improvements in linker design and recent breakthrough data (e.g. see Padcev + Keytruda, Enhertu…).
M&A activity has significantly increased: 2020 (13), 2021 (17), 2022 (40) and 2023 (41). A few notable examples from 2023:
Merck / Daiichi (licensing): $4B upfront, $22B total value to license 3 ADCs
Pfizer / Seagen (acquisition): $43B acquisition (Seagen has 4 approved ADCs and a rich pipeline)
GSK / Hansoh (licensing): $85M upfront, $1.5B milestones for one ADC
BioNTech / DualityBio (licensing): $170M upfront, $1.5B total value, license 2 ADCs
Lilly / Mablink (acquisition): undisclosed, Lilly / Emergence (acquisition): est $800M (upfront + milestones)
AstraZeneca / KYM Biosciences (licensing): $63M upfront, $1.1B total, AstraZeneca / LaNova Medicines (licensing): total $600M
Aiolos Bio raises $245M Series A for an old-fashioned biotech business model
How to start an old-school biotech:
Be a pharma veteran and find a pal who is too (Aiolos: Genentech)
Make a list of compelling immunology targets (Aiolos: TSLP)
Scout developing / under-appreciated assets against these targets (particularly in China) that have decent patient data and a differentiation factor over competitors (Aiolos: Jiangsu Hengrui, 100 patients cohort data, 6 month dosing regimen over monthly)
License the asset (Aiolos: $21M upfront, back-ended milestones based on success)
Raise lots of money and take to clinic
Leading biotech investor OrbiMed raises $4.3B fund
Despite the bearish public market, OrbiMed sees opportunities in biotechs that have a (1) relatively low burn (2) focus on later-stage assets (3) strong pharma partnership strategy.

What we liked on Twitter


Events


Field Trip
Did we miss anything? Would you like to contribute to Decoding Bio by writing a guest post? Drop us a note here or chat with us on Twitter: @ameekapadia @ketanyerneni @morgancheatham @pablolubroth @patricksmalone @ZahraKhwaja