

BioByte 39: the biomolecular atlas, SOTA deep learning for RNA structure prediction, applications of scRNA in drug development, AI x Bio Summit
BioByte 39: the biomolecular atlas, SOTA deep learning for RNA structure prediction, applications of scRNA in drug development, AI x Bio Summit




Welcome to Decoding Bio, a writing collective focused on the latest scientific advancements, news, and people building at the intersection of tech x bio. If you’d like to connect or collaborate, please shoot us a note here or chat with us on Twitter: @ameekapadia @ketanyerneni @morgancheatham @pablolubroth @patricksmalone. Happy decoding!

In a hurry? Here’s what we read this week:
A primer on LLMs in Computational Biology
Chris Gibson’s secret to building Recursion Pharma (hint: its all about people)
Frontotemporal Dementia’s devastating impact
Human BioMolecular Atlas Program (HuBMAP): a global initiative that aims to assemble spatial maps of biomolecules, including RNA, proteins, and metabolites, in human organs at single-cell resolution.
A new model achieves SOTA status in predicting RNA secondary structure
Practical applications of scRNA sequencing in drug discovery and development
CellRank2, a versatile and scalable framework for studying cellular fate using multiview single-cell data

What we read
Blogs
Large Language Models in Computational Biology – A Primer [Jian Ma, Carnegie Mellon]
Not a blog post, but rather the slides from a talk given by Jian Ma, a Professor of Computational Biology at Carnegie Mellon. The most relevant slides for those interested in comp bio begin on slide 14, and provide a great overview of recent LLMs applied to genomic sequence data (eg DNABERT-2 and Nucleotide Transformer) and single-cell data (eg scGPT and Geneformer). The talk concludes with a list of open questions for the field (slide below). A few additional research topics we’re thinking about:
prompt engineering for biology (output of LLMs depend heavily on prompt)
how best to incorporate human scientists in the training loop (eg RLHF)
what is the best pre-training task for genomics

How Chris Gibson founded Recursion Pharmaceuticals [Chris Gibson, Utah Businesswire, July 2023]
“In many ways, that failure reflected a problem that plagues our entire industry: We were limited by the tools at our disposal, leading us to a reductionist view of biology and a hypothesis that was inherently biased.”
Chris describes the origins of the idea for recursion: using new tools to expand our view of biology and to discover medicines at a scale and pace unlike any other using technology. His drive to tackle complex, hard problems came from sixth-grade – where he chose a challenging scientific project but missed the mark. His teacher taught him that it's better to “fail at something difficult, rather than succeed at something easy”.
Being one of the first companies of its kind, Recursion had to define a new culture. Chris mentions that the life sciences “is set up in a way that teaches people to build upon prior work and avoid challenging dogma”, which is very different from the culture of fields like data science “where you’re taught to challenge assumptions wherever possible”. Finding people who are experienced but also believe there is a better way of approaching drug discovery is challenging, and in many ways people and culture is harder than the science he explains. Dig into the full article for some deeper insights about Recursion’s culture.
The Vanishing Family [Robert Kolker, New York Times, July 2023]
Kolker writes a captivating piece about life in a family afflicted by an autosomal dominant genetic disease—frontotemporal dementia (FTD). We’ve written about FTD in the context of Modulo, a neuro startup working to create treatments to the disease, but Kolker’s narrative paints a gripping picture of what life is like when your loved ones are “disappearing into dementia in middle age”, not knowing if you will meet the same fate in the next several years. Kolker walks through how difficult it can be to get a diagnosis for rare diseases including FTD where there is no simple test. Genetic analysis can still leave you in the dark, as very specific parts of the genome need to be sequenced, analysis which is often impossible to get access to. It’s a chilling tale that delves into the sacrifices patients (and potential patients) make in their daily lives leading up to diagnosis and after. It’s a tale into what life is like as a caretaker and as a patient. Highly recommend the read if your work interfaces with patients of any kind—we can all use the reminder of what a blessing good health is and what we are all working towards solving.
Human BioMolecular Atlas Program [Nature Portfolio, July 2023]
Advances in molecular biology have generated a wealth of information about the components of individual cells in the human body. However, it is still unclear how these components function together. The Human BioMolecular Atlas Program (HuBMAP) is a global initiative that aims to assemble spatial maps of biomolecules, including RNA, proteins, and metabolites, in human organs at single-cell resolution. The ultimate goal of HuBMAP is to enhance our understanding of how cells work and how they interact with each other in the human body. More than 60 institutions and 400 researchers around the world are participating in the initiative.
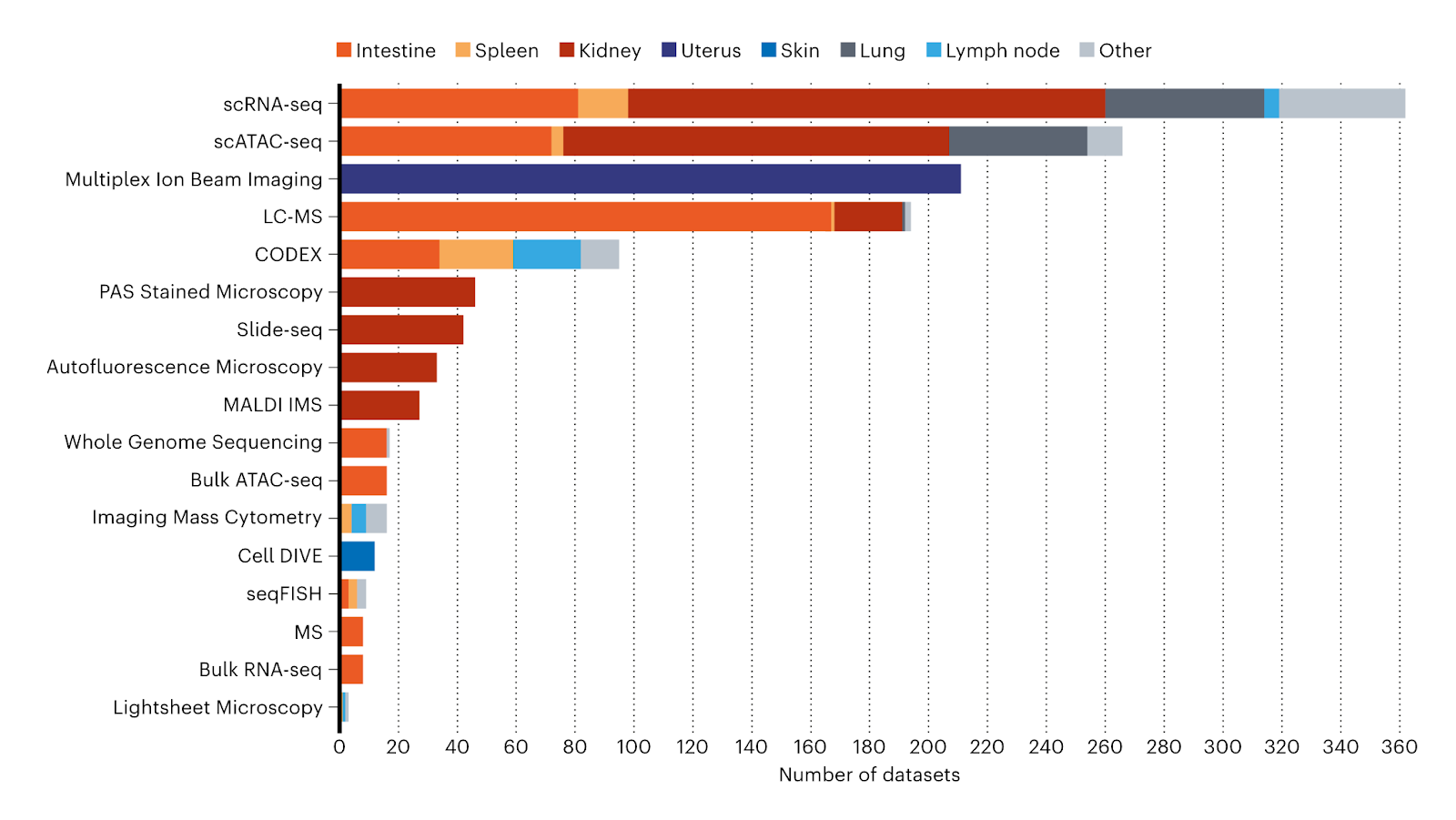
The HuBMAP consortium has developed methods, tools, and standards for collecting and analyzing data. These advancements enable the integration of data across various organs by research groups and consortia. By using these methods, valuable information about the transcriptome, epigenome, proteome, and metabolome across different human tissues and organs has been revealed. These findings cover a wide range of spatial scales, from subcellular resolution at approximately 100 nanometers to organ-level measurements at the centimeter scale.
All the data collected by the HuBMAP consortium are freely available on the HuBMAP Data Portal.
Academic papers
Scalable Deep Learning for RNA Secondary Structure Prediction [Franke et al., arXiv, 2023]
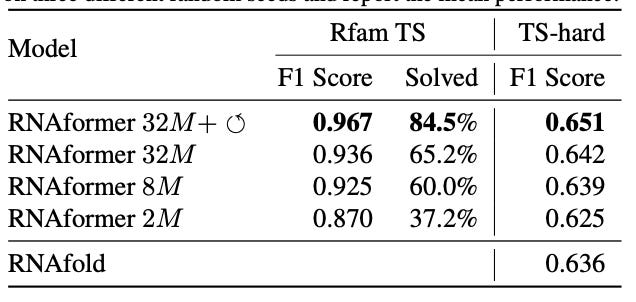
Why it matters: RNA function is dependent on the tertiary structure of RNA which in turn is dependent on the secondary structures of RNA. The secondary structure of RNA describes the intramolecular base-pair interactions determined by the sequence of nucleotides. Secondary structures can guide the design of RNA–based and RNA-targeting therapeutics, making their accurate prediction a desirable problem to solve.Franke at al. present the AlphaFold-inspired RNAformer, a deep learning model using Transformer-like axial attention and recycling in the latent space to gain performance improvements to predict RNA secondary structure from sequence. It achieved SOTA performance on the TS0 benchmark dataset.
Axial attention captures dependencies between positions along a specific axis of input data. In the case of RNAformer, axial attention is applied to create a dependency between all potential nucleotide pairings. Recycling of the processed latent space increases the model depth, which allows the model to reprocess and correct its own predictions.
Loss of TJP1 disrupts gastrulation patterning and increases differentiation toward the germ cell lineage in human pluripotent stem cells [Vasic et. al, Developmental Cell, June 2023]
Why it matters: as far as science has come, little is known about early development and how embryos grow. This paper examines the role that tight junctions between cells play in gastrulation to influence cell fate and differentiation. The more we know about the processes and signaling pathways that regulate embryo formation, the more readily we can repeat these processes in a controlled environment. The paper shows a repeatable way to study gastrulation, the fundamental process of cells forming the three main layers of cells found in the embryo once exposed to signaling factor BMP4. The authors found that though all cells are exposed to BMP4, varying levels making it to the individual cell type is responsible for which of the three layers that cell goes on to form. This is modulated through tight junctions, allowing BMP4 to give different cues to different cells depending on location.
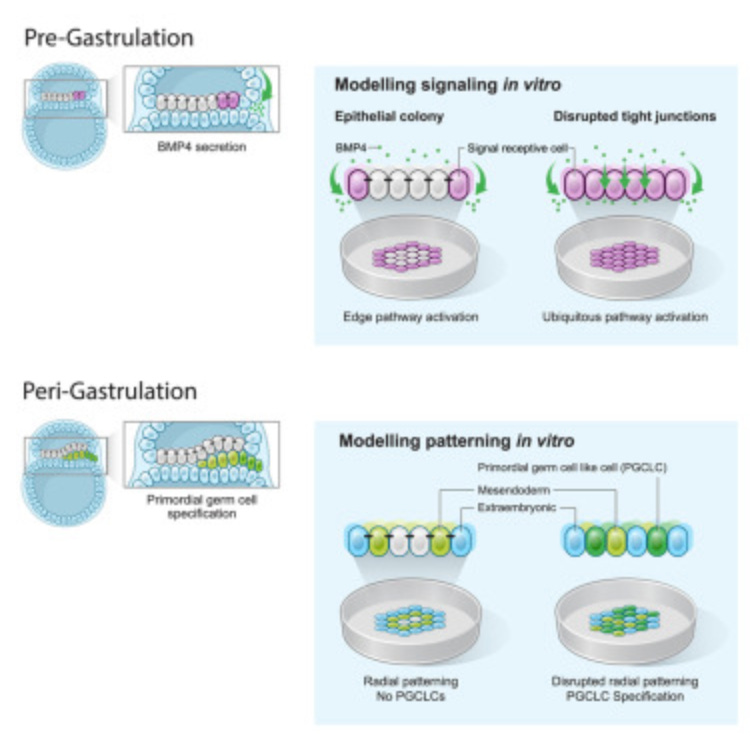
Using CRISPR, the authors knocked out production of TJP1, a protein that is partially responsible for forming tight junctions in iPSCs. BMP4 activated all cells in that group highlighting the importance of these tight junctions. It’s long been a challenge to generate consistent homogenous populations of differentiated stem cells. Looking towards modulating tight junctions as this study has shown us, may hold part of the solution.
Applications of single-cell RNA sequencing in drug discovery and development [Van de Sande et al., Nature Reviews Drug Discovery, June 2023]
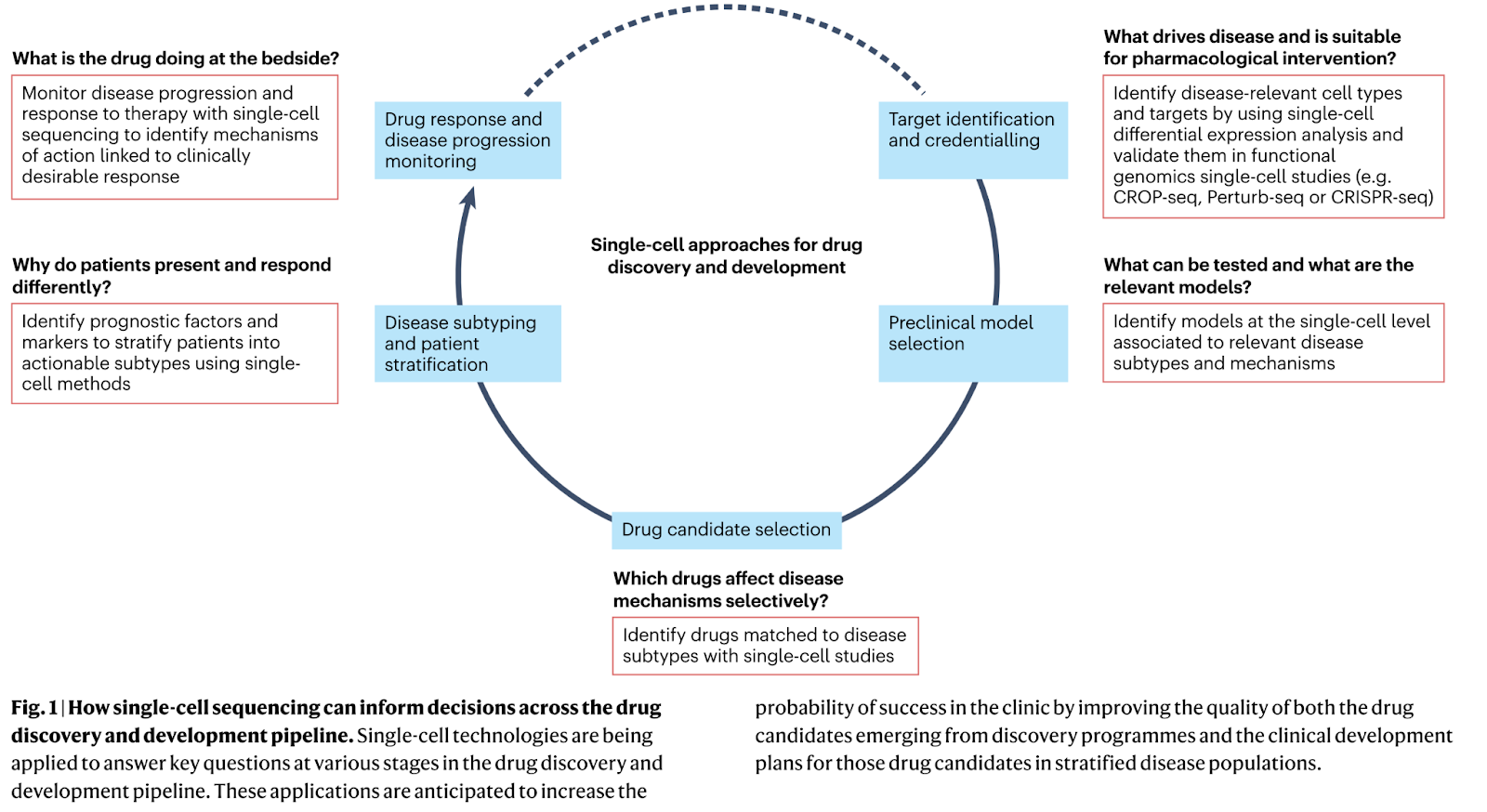
Why it matters: Single-cell RNA sequencing (scRNA-seq) is a powerful technique that allows researchers to analyze the gene expression profiles of individual cells. This method provides a high-resolution view of cellular heterogeneity and can reveal previously undiscovered cell subpopulations, biomarkers, and drug resistance pathways. This review, published by a number of researchers from biopharma organizations like Sanofi, GSK, Moderna, and others, overviews some of the most important applications of scRNA-seq in drug discovery including:
Disease understanding: scRNA-seq enables the identification of distinct cell subtypes within a tissue, leading to a better understanding of disease mechanisms and the identification of potential therapeutic targets. For example, single cell profiling of human dopamine neurons in Parkinson’s disease identified ten transcriptionally defined dopaminergic subpopulations in the substantia nigra, but only one population selectively degenerates.
Target discovery, credentialing, and validation: Multiplexed functional genomics screens incorporating scRNA-seq can enhance target credentialing and prioritization. For example, a novel cell-type specific immunotherapy target (S100A4) in glioblastoma was identified using single cell analysis of glioma samples.
Drug screening and MoA analysis: high-throughput screening of chemical libraries can be combined with single cell gene expression readouts to help elucidate a drug’s MoA. For example, the gene expression profiles induced by drugs with known MoAs can be compared to a drug of unknown MoA.
The review concludes with a number of current challenges in the field, including:
Study design and implementation: there is still massive heterogeneity in study design across published experiments. A uniform analysis pipeline and more consistent methodology is needed.
Data accessibility: uniform data aggregation and metadata is improving (eg the Broad Single Cell Portal), but overall still lacking.
Data interoperability and reusability: large variability in the format of publicly available data, as well as inconsistent QC and lack of well-defined cell type nomenclature.
Unified fate mapping in multiview single-cell data [Weiler, et al., BioXriv, July 2023]
Why it matters: Single-cell RNA sequencing (scRNA-seq) is a powerful tool for studying cellular heterogeneity and differentiation. However, traditional scRNA-seq analysis methods fail to account for additional information that can be informative about cellular state changes, such as experimental time points, multimodal measurements, and metabolic labeling data.Following our past coverage of cellular fate mapping, a new pre-print details the launch of CellRank2, a versatile and scalable framework for studying cellular fate using multiview single-cell data.
The CellRank 2 framework addresses the challenge of incorporating multiple data modalities into trajectory inference by decomposing the problem into two components:
Modality-specific modeling of cell transitions: models the transitions between cell states for each data modality separately. This is done by developing a kernel that captures the similarity between cell states in each modality.
Modality-agnostic trajectory inference: combines the kernels from the modality-specific modeling step to infer trajectories that are consistent across all data modalities. This is done by using a Markov chain model to represent the probability of transitioning from one cell state to another.
CellRank2 can be used to analyze multiview data from millions of cells and has been shown to be effective in a variety of applications, including human hematopoiesis, mouse endodermal development, and mouse intestinal organoid differentiation.
One of the key features of CellRank2 is its ability to incorporate experimental time points into trajectory inference. This is done by developing an efficient optimal-transport-based kernel that allows for the simultaneous modeling of inter- and intra-time point information. This approach has been shown to be effective in recovering terminal states more faithfully, allowing for the study of gene expression change continuously across time, and discovering putative progenitors missed by alternative approaches.
CellRank2 is also capable of deciphering cellular dynamics from metabolic labeling data. This is done by introducing a new computational approach that recovers cell-specific transcription and degradation rates. This information can then be used to study gene regulatory strategies underlying cellular state changes.
Overall, this work suggests that CellRank2 has the potential to be a valuable tool for understanding and conceptualizing fate choice as single-cell datasets grow in scale and diversity.
What we listened to
Notable Deals
Cell therapy startup Turnstone adds to biotech IPO flurry with $80M raise
Riding on positive data, argenx raises $1.1B while Alzheimer's biotech Acumen lands $130M
A-Alpha Bio, a biotech startup that measures and predicts protein interactions, raises $22.4M
In case you missed it
Creating new biology using AI [Ali Madani, The Naked Scientists, 2023]

What we liked on Twitter
Why did $NVDA choose $RXRX Recursion as its techbio bet? @PierceARK
Events
Thanks to everyone who came to the first edition of the AI x Bio Summit at the NYSE.

Field Trip
Did we miss anything? Would you like to contribute to Decoding Bio by writing a guest post? Drop us a note here or chat with us on Twitter: @ameekapadia @ketanyerneni @morgancheatham @pablolubroth @patricksmalone